
Step 1: Find Your Login URL
https://office.com/signin – This link will take you directly to the Office 365 sign-in.
https://myapps.microsoft.com/[your domain].com – You can log in via the MyApps URL, and if corporate branding is applied to the login pages for your organization, you can add your domain to the URL as shown to load the branded version of the page.
https://www.office.com – This link will take you to the main Microsoft Office landing page. On this page (and others throughout office.com and microsoft.com) you will find a “Sign in” link at the top of the page. Click the link to sign in.
https://[yourcompanysubdomain].sharepoint.com – If your organization has an Office 365 license that includes SharePoint, you can go directly to your organization-specific URL and log in there. Contact your internal IT department or network support team to obtain the URL.
A quick note about corporate branding. If your company has branded your sign-in page, you will not see the branded experience at first when going through the generic office.com or microsoft.com sign-in pages. However, once you enter your email address and click continue, the page will refresh and you will see your company’s branded sign-in page upon being prompted to enter your password.
Step 2: Sign In With Your Credentials
The next step is to sign in with your user credentials.
Microsoft Office 365
 |
Using a web browser, visit the URL from step 1. Enter your username in the field provided and click Next. Typically, your username is your company email address. |
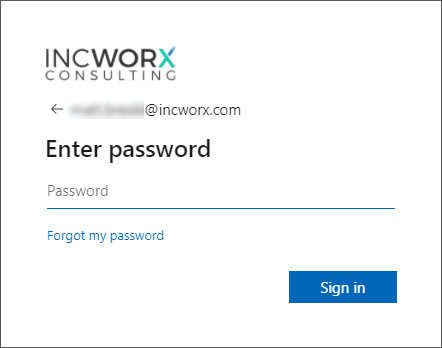 |
Enter your Office 365 password and click Sign in.
Notice the presence of the IncWorx logo included in the sample image of the sign-in page. This is an example of an Office 365 sign-in page where corporate branding has been applied. Once Office 365 recognized the domain to which you are attempting to gain access, it renders the appropriate company’s page. |
On-Premises (hosted locally by your organization or hosted by a third-party)
 |
Using a web browser, go to your company’s SharePoint site using the URL from step 1.
Depending on how your company’s login page is configured, you may get prompted for credentials from a web-based form, or via a traditional windows login prompt similar to the image. In either case, log in using your company network credentials. Typically, your username will be in the format of either companydomain\jdoe or [email protected]. |
Tips & Troubleshooting: Forgot Password
Microsoft Office 365
If you’ve forgotten your Office 365 login credentials, Microsoft provides an automated solution for resetting your password here: https://passwordreset.microsoftonline.com.
On-Premises (hosted locally by your organization)
If you’ve forgotten your corporate network credentials, you will need to contact your internal help desk or network support team. They will be able to reset your password and supply you with a temporary password.
Hosted by a Third-Party
If you’ve forgotten your credentials, they may be managed by either your internal IT department or your third-party hosting provider. Contact your internal help desk or network support team first. If neither exists, you may need to contact the third-party’s support department to request your password be reset. You should be able to find a phone number, email address, or even live chat on the third-party’s site to begin your request for a password reset.


